20. Árboles de Especies, árboles de Genes y Clasificación del Linaje incompleta
19 de Julio de 2013. Temas: Genética
Nota: Esta serie de artículos ha sido concebida como una introducción básica a la ciencia de la evolución para no especialistas. Aquí se puede ver la introducción a esta serie o volver al índice aquí.
En este artículo discutimos de qué manera se espera que la distribución de algunos alelos de especies relacionadas no coincida en el conjunto del árbol filogenético de la especie.
En los últimos artículos de esta serie hemos examinado el patrón general que observamos al comparar los genomas relacionados entre sí, y cómo distintos conjuntos de datos coinciden en el mismo árbol filogenético, o filogenia. En este artículo, trataremos de comprender con más profundidad las filogenias, y cómo es que en realidad sí se espera que algunas características de los genomas no concuerden con sus árboles filogenéticos.
Pero antes que nada, un breve comentario: éste es un tema difícil, que podría resultar confuso al principio. Pero si hemos llegado hasta aquí leyendo esta serie de artículos, tenemos ya las herramientas necesarias para entender qué es lo que está pasando aquí y, con un pequeño esfuerzo adicional, lograremos una comprensión todavía más profunda de los genomas relacionados, que la que teníamos hasta ahora. Si, por otra parte, este tema en concreto parece un poco lioso, no nos preocupemos: el resto de la serie no depende de la comprensión de esto con detalle. También, por supuesto, conviene preguntar, en los comentarios, si las cosas no quedan claras.
Árboles de especies
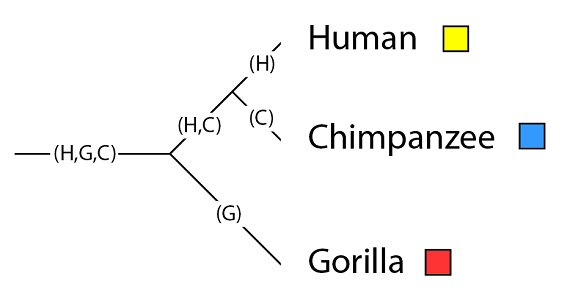
Volvamos al ejemplo de la filogenia, ya familiar a estas alturas, de los humanos, chimpancés y gorilas:
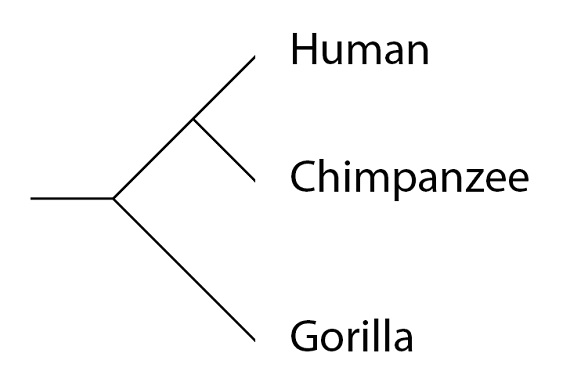
Las “filogenias” se conocen también como “árboles de especies” o “árboles filogenéticos de especies”. Un árbol filogenético de una especie nos muestra el patrón general; qué especies comparten una población ancestral común más recientemente y cuáles la comparten en un pasado más lejano. En otras palabras, como hemos señalado en el último artículo de esta serie, una filogenia es una medida de la historia, compartida o separada, para cualquier par de especies. Cuanto más larga sea la historia común entre dos especies, más parecidas esperamos que sean, por término medio. Humanos y chimpancés, por ejemplo, siguieron compartiendo una historia común durante varios millones de años, después de que el linaje del gorila se separara de la población ancestral común, antecesora tanto del hombre como del chimpancé. Esta historia compartida es lo que, por término medio, hace que los genomas humano y del chimpancé sean más parecidos entre sí que lo que se parecen cualquiera de ellos al genoma del gorila. Los genes individuales, y sus alelos, pueden tener una historia diferente en cada especie, al separarse ésta de las otras. Para este tipo de análisis tenemos que examinar las filogenias de los genes individuales, los llamados “árboles de genes”.
Árboles de genes
Si comprendimos en artículos anteriores (“El fundamento de la variación heredable, Parte 1 y Parte 2”) el modo en que la variación (los alelos) surge por mutación, debería resultar bastante intuitivo que los mismos principios que pueden utilizarse para agrupar las especies en una filogenia pueden también utilizarse para agrupar los alelos de un mismo gen en otra filogenia. Por ejemplo, consideremos la secuencia de ADN de tres alelos del mismo gen, que podemos representar como alelos “amarillo”, “rojo” y “azul” (los cuadros coloreados). Las diferencias en la secuencia que hacen distintos a esos alelos se destacan en rojo:
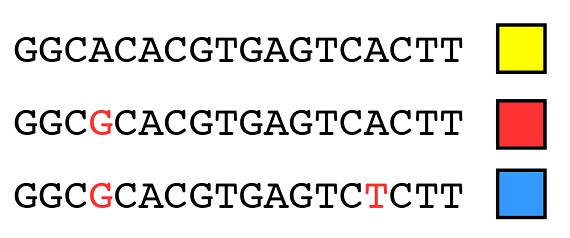
Con los mismos principios que hemos utilizado para las especies en conjunto podemos explicar el origen de estos tres alelos por medio de dos mutaciones, empezando con el dato de que el alelo amarillo es el estado ancestral:
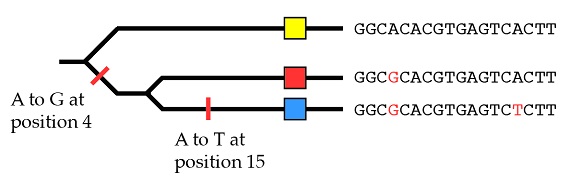
Así que, dentro de una población, podemos reconstruir la historia de los alelos de un gen individual utilizando los mismos métodos que habíamos aplicado previamente a las especies en su conjunto.
La especiación junto con la variación genética a lo largo del viaje (o no)
Así pues, la mutación está constantemente produciendo alelos nuevos, es decir variación, dentro de las poblaciones, y los procesos tales como la selección natural y la deriva genética trabajan conjuntamente para o bien aumentar o bien disminuir la frecuencia de los alelos en las poblaciones, a lo largo del tiempo.
También hemos empleado bastante tiempo en discutir (en los cuatro artículos titulados “De la variación a la especiación” Parte 1, Parte 2, Parte 3 y Parte 4) cómo tiene lugar la especiación, empezando con poblaciones que se separan unas de otras y que, a lo largo del tiempo, acumulan diferencias que pueden conducir a la formación de especies distintas. Lo único que falta ahora es reunir todas estas ideas: considerar lo que podría suceder con la variación (los alelos), dentro de una población a medida que ésta va sufriendo la especiación. Para hacerlo, rastreemos a nuestro hipotéticos alelos a través de la especiación que conduce a la especie humana, a los chimpancés y a los gorilas.
Este árbol filogenético de especies tiene las siguientes poblaciones: la población que es ancestral a las tres especies, denominada como “(H, G, C)”, de “(Hombre, Gorila, Chimpancé)”; la población ancestral común de humanos y chimpancés (H, C), y los linajes (las poblaciones) que llegan a las especies en la situación actual tras su última especiación, con las especies en la filogenia (H), (G) y (C):
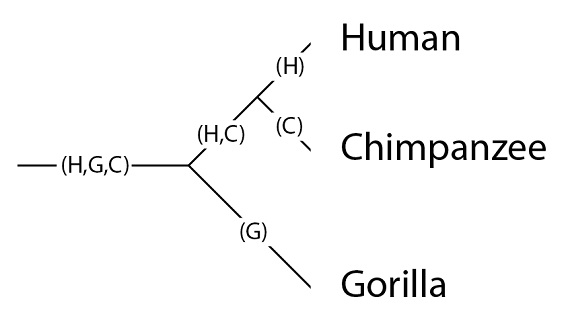
Es importante tener en cuenta que una simple línea, en la filogenia, es en realidad una población, y que las poblaciones pueden tener variación genética. Situemos nuestros tres alelos en la población (H, G, C):
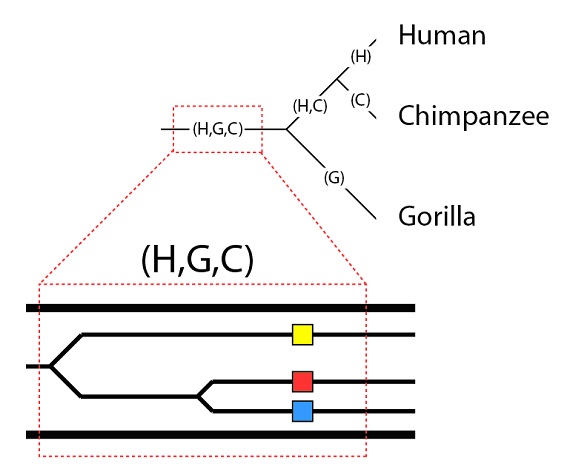
Pasamos ahora a explorar las posibilidades de cómo estos alelos se van a heredar, o no, a lo largo de la especiación que va a tener lugar. Una posibilidad es directa: los tres alelos se heredarán en las tres especies. Esta es la posibilidad llamada de “clasificación completa del linaje”, porque representa una segregación completa de todos los alelos en los tres linajes. Esto requiere que los tres alelos estén presentes en las subpoblaciones que se dividen en los distintos linajes, y que no se pierda ningún alelo, con el tiempo, en ninguno de los linajes. Aunque esto es, desde luego posible, no quiere decir que sea incuestionable. Como hemos visto, cuando las poblaciones se separan es improbable que todos los alelos de la población original queden representados en las dos subpoblaciones tras la división. Además, es posible que la selección o la deriva genética puedan, con el tiempo, provocar la pérdida de alelos en uno de los linajes pero no en el otro. Todo lo que no sea una perfecta segregación de todos los alelos se llama “clasificación del linaje incompleta”; y para un genoma grande es un hecho que al menos algunos de los genes sí mostrarán este efecto.
Clasificación del linaje incompleta; un ejemplo
La primera dificultad que encuentran estos tres alelos para la clasificación completa del linaje es el proceso de especiación al que se enfrentan, que separa los linajes (H, C) y (G). Para los propósitos de este ejemplo, supongamos que el alelo rojo sea excluido de la población que forma el linaje (H, C), pero que los tres alelos permanezcan en el linaje (G). Recordaremos que éste es un ejemplo del efecto fundador- una muestra no aleatoria que puede excluir alelos de una subpoblación nueva por azar:
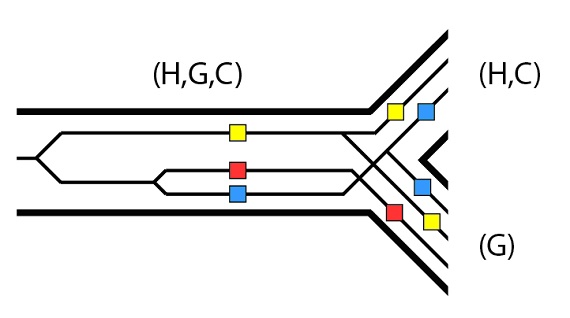
Pues ahora veamos un posible escenario que se sigue del proceso de especiación (H, C) / (G). En el linaje (G), los alelos amarillo y azul se pierden con el tiempo. En la especiación (H) / (C), tanto el alelo azul como el amarillo se segregan en ambos linajes, pero en el linaje (C) el alelo amarillo acaba por perderse con el tiempo. Lo mismo sucede con el alelo azul en el linaje (H), que acaba perdiéndose más tarde:
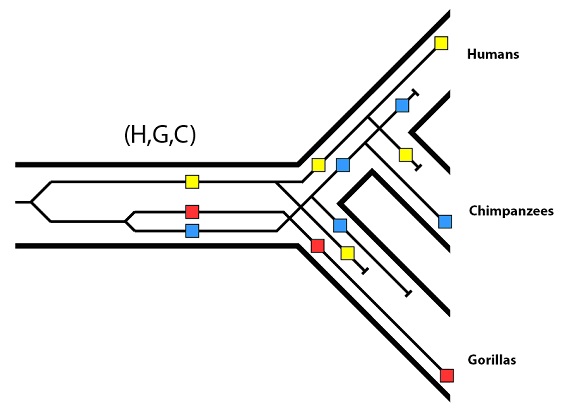
Luego para este gen en particular tenemos, entonces, el patrón siguiente:
Y al final lo que vemos es esto: el árbol de los genes de estos tres alelos no concuerda con el árbol filogenético de las especies. Recordemos que en el árbol de genes, los alelos rojo y azul están más estrechamente relacionados entre sí de lo que lo están con el amarillo.
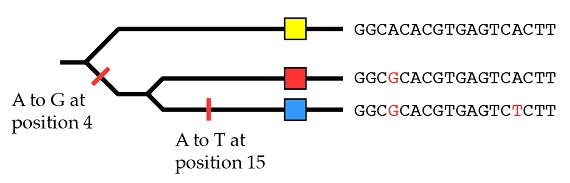
En el árbol de especies, sin embargo, los dos parientes más cercanos, chimpancés y humanos, no tienen los alelos más cercanos: tienen los alelos más distantes entre sí.
Ahora que hemos elaborado este ejemplo, es de esperar que nos quedará clara la razón de esta discrepancia: no hay garantía de que los alelos se ordenen en un linaje de forma que coincidan con el patrón general de las especies. Si un gen muestra variación en una población en proceso de especiación podemos esperar que en ocasiones se ordene con un patrón que no coincida con el patrón de las especies; en ciertos casos habrá un árbol de genes que será “discordante” con el árbol filogenético de las especies. En una población con miles de genes con múltiples alelos, es un hecho que algunos alelos se van a ordenar con un patrón discordante. Lejos de ser esto un problema para la evolución, los árboles discordantes son predichos por la evolución. Sería un problema el que no los encontráramos, pero de hecho sí lo hacemos y, como veremos más adelante, los observamos precisamente en el patrón que concuerda con lo que esperaríamos basándonos en los árboles filogenéticos de las especies.
En el siguiente artículo de esta serie, discutiremos cómo los árboles de genes discordantes pueden utilizarse para determinar otra característica interesante para los científicos: los tamaños de población de los linajes en una filogenia.


